Raman spectroscopy is widely applied in identifying local structures in materials, but the interpretation of Raman spectra is non-trivial. An accurate computational database of reference spectra calculated with a consistent level of theory can significantly aid in interpreting measured Raman spectra. Here, we present a database of Raman spectra of inorganic compounds calculated with accurate hybrid functionals in density functional theory. Raman spectra were obtained by calculating dynamical matrices and polarizability tensors for structures from the Inorganic Crystal Structure Database. The calculated Raman spectra and other phonon properties (e.g., infrared spectra) are stored in a MongoDB database publicly shared through a web application. We assess the accuracy of our Raman calculations by statistically comparing ~80 calculated spectra with an existing experimental Raman database. To date, the database contains 161 compounds and is continuously growing as we add more materials computed with our automated workflow.
Raman scattering is the inelastic scattering of light by either absorbing or creating lattice vibrations (phonons) 1 . Raman spectroscopy provides local structure fingerprints of materials by detecting changes in the frequency of incident monochromatic radiation due to the scattering 2,3 . Materials identification with Raman spectroscopy holds advantages including high chemical sensitivity, tolerance to long-range disorder, and high spatial resolution 4,5 . The non-invasive and non-destructive Raman measurement sees applications in in-situ and operando characterization modes 6 . Compared to other phonon measurement techniques, such as neutron scattering, Raman spectroscopy offers better frequency resolution, easy sample preparation, relatively inexpensive equipment and modularity of lasers, and its fast operation 3,4,5 . Thanks to these advantages, Raman spectroscopy is widely used in the characterization of biological and organic molecules, as well as inorganic materials in various fields including energy storage and conversion 6,7,8,9,10,11 , catalysis 12,13,14 , low-dimensional materials 15,16 , biomedical applications 17,18,19 , and others. However, the interpretation of experimental Raman spectra may be complicated requiring significant time and effort. This urges the need for accurate computational references to improve the speed and accuracy of Raman spectra’s interpretation.
First-principles calculations of lattice dynamics and phonons have gained considerable attention and are pushing forward the sub-field of computational Raman spectroscopy 20,21,22 . In 2015, Togo and Tanaka built the well-known first-principles phonon database that contains the full phonon dispersion of a large number of inorganic and ordered materials 23 . This database showcases the power of phonopy -a software package developed to predict the phonon properties of ordered crystals in combination with the accuracy enabled by first-principles calculations 24 . More recently, Liang and co-workers reported a database of computed Raman spectra of approximately 55 compounds derived from density-functional perturbation theory (DFPT) calculations and finite difference derivative of the dielectric tensors 25 . Taghizadeh et al. applied time-dependent third-order perturbation theory to calculate Raman spectra of 733 two-dimensional (2D) monolayer materials and used the calculated spectra to identify materials from experimental Raman spectra 26 . Popov and co-workers proposed a new average method for computing Raman spectra of polar polycrystalline materials from DFPT 27 . Most recently, utilizing force constant matrices from the Togo phonon database, Bagheri et al. built a Raman database by only calculating Raman tensors by finite differences 28 .
This important progress represents the “bleeding edge” of computational Raman spectroscopy for the purpose of spectra interpretation and further data-driven development. However, one common shortcoming of all these databases is that the predictions are limited to rather low levels of theory, namely the generalized gradient approximation (GGA) exchange-correlation functionals 29,30 , while more accurate hybrid functionals (beyond “plain vanilla” GGA) have not been applied in calculating Raman databases. In particular, the 2D Raman library by Taghizadeh et al. is based on the Perdew–Burke–Ernzerhof (PBE) functional 26 , Liang and co-workers’ database was calculated using PBE + U 25,31,32 , and the database by Bagheri et al. are based on the revised PBE for solids (PBEsol) 28,33 . All these predictions are plagued to a different degree by the pernicious self-interaction error of the semi-local GGA functionals. However, the use of such low-level theories is due to the intrinsic complexity and high computational cost of phonon calculations with higher-level theories, and in particular, their implementation in plane-wave and pseudo-potential codes. Therefore, an efficient and accurate hybrid-functional computational Raman database is in high demand.
In the present data records, we compute Raman spectra together with phonon properties at a consistently high level of theory, namely the hybrid functional PBE0 34 , in a linear combination of atomic orbitals, expanded in terms of triple-ζ valence with polarization (pob-TZVP-rev2) Gaussian basis sets 35,36 . These approximations are realized using the CRYSTAL code at a relatively low computation cost thanks to its exploitation of lattice symmetry during frequency calculations 37,38,39 . Here, the computation of Raman spectra is automated such that experimentally measured structures are “digested” into the CRYSTAL code, followed by the calculation of Raman spectra and other phonon properties, which are subsequently stored into a MongoDB database. The database is interfaced with an interactive user-friendly web application.
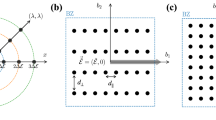
A small fraction of an incident light beam is scattered when it passes through a substance, or more precisely a material. The inelastic part of the scattering is known as Raman scattering, which changes its frequency by creating or absorbing phonons. From group-theory analysis of crystal symmetry, only particular phonon modes are allowed to take part in Raman scattering. The intensity of the scattering is dependent on the polarizability of materials because the scattering originates from the polarization fluctuations induced by the electric field of the incident electromagnetic radiation. Frequencies of the Raman-active phonon modes and the scattering intensities are calculated to get Raman spectra for inorganic compounds.
Our calculations of Raman spectra are performed using the ab initio computational program CRYSTAL 37,38,39 . The calculations with CRYSTAL have several unique features that facilitate efficient and accurate computation of lattice dynamics: (i) In contrast to most commonly used plane–wave basis sets, the crystalline orbitals are expanded as linear combination of atom centered atomic–orbitals, (ii) the hybrid functional PBE0, with 25% Hartree–Fock exchange 34 , is used rather than the semi-local GGA functionals, such as PBE 30 , and (iii) point group symmetry of lattice is utilized at multiple levels (i.e., atomic positions, wave functions, etc.) during the calculations of vibration frequencies, which means the calculations are only performed on symmetrically irreducible atoms.
Vibration frequencies are determined in CRYSTAL by calculating and diagonalizing mass-weighted dynamical matrices (bottom box in Fig. 1) 40,41 . The dynamical matrix is a 3n × 3n (n = number of atoms, 3n is the number of vibrational normal modes) matrix with components being the second partial derivatives of the DFT total energies versus positional displacements, which are calculated by numerical differentiation of the analytical gradient of the DFT total energy with respect to the atomic positions. Diagonalization of an appropriately symmetrized mass-weighted dynamical matrix then gives the normal modes (eigenvectors) and vibration frequencies (eigenvalues).
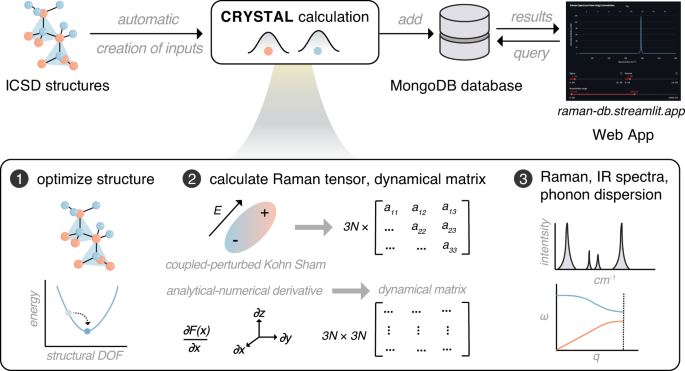
In CRYSTAL, Raman intensities are calculated directly through the Raman polarizability tensors obtained via perturbation theory, i.e. the coupled-perturbed Kohn-Sham (CPKS) approach (middle box in Fig. 1) 42,43,44,45,46 . In this approach, a static electric field is applied along different directions to calculate the first-, the second-, and the third-order electric susceptibility tensors, which can be converted into polarizability and dielectric tensors, the first hyperpolarizability tensor, and second hyperpolarizability tensor, respectively. From the electric susceptibility tensors, a 3 × 3 Raman tensors are calculated for each of the 3n normal modes. The Raman intensities can then be calculated using the following equations:
where Iij(n) is the directional Raman intensity for single crystals, X(n, i, j) is the i, j-th component of the Raman tensor expressed in the basis of normal mode n, and V is cell volume. Placzek rotation invariants 5 are used for averaging the mode Raman tensors of single crystals with Eqs. 2–4: